Modeling virus actions & inhibition
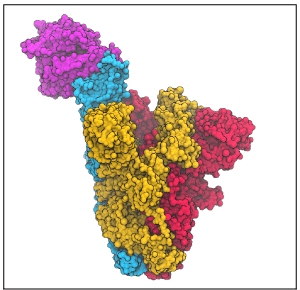
- How the virus enters the cell: Carlos Simmerling models how the SARS-CoV-2 spike protein Receptor Binding Domain (RBD) binds to the host cell’s ACE-2 receptor, with the aim of drug discovery. Starting from cryo-EM structures in Cell and Science, he reconstructs missing details and loops using BRIKARD (Coutsias), then performs MD and pathway computations to explore the protein’s motions and conformational changes.
- The structures of the viral proteins: The structures of most COVID virus proteins are not yet known. The Dill group is computing those structures using MELD x MD, and testing in CASP 14. And the Kozakov group is computing structures of human-virus and virus-virus protein pairs.
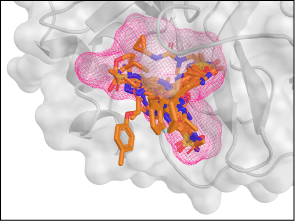
- Small molecule inhibitors of the viral protease: The Kozakov, Rizzo, and Dill groups seek inhibitors of the SARS-CoV-2 proteases that are necessary for viral reproduction. First, they perform rapid virtual screening of possible binders from a large database using DOCK and our template-based LigTBM, followed by atomistic physics Monte Carlo and MELD x MD.
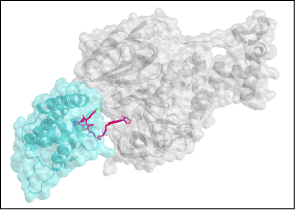
- A PROTAC linker to degrade viral proteins: The Kozakov and Dill groups are designing a PROTAC linker (PROteolysis-TArgeting Chimeras) that hooks a viral protein onto a host cell’s ubiquitin protein to send the viral protein to the cell’s `garbage disposal’ proteasome system. They use CLUSPRO protein docking software with atomistic refinement by MELD x MD. Peter Tonge’s group at SBU is making and testing the linkers.
