AMBERsb (Simmerling)
Molecular simulations with solvation models & forcefield
With over 10,000 citations, Simmerling’s energy functions are widely regarded as the most accurate model available for protein dynamics. Simmerling’s Amber ff99SB sets a new standard for protein force-field accuracy, continued in ff14SB and ff19SB.
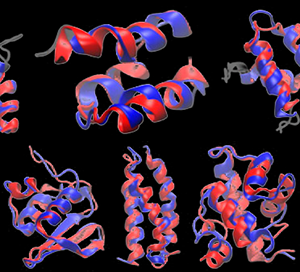
Simmerling published one of the first successful simulations using atomic-detailed dynamics simulations to predict an accurate structure for a small protein.
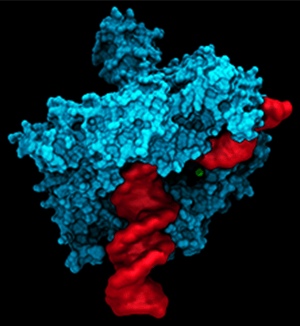
Amber simulations show how DNA repair enzymes can recognize damaged bases in the sea of normal DNA, bind to them with striking specificity, and repair the damage.
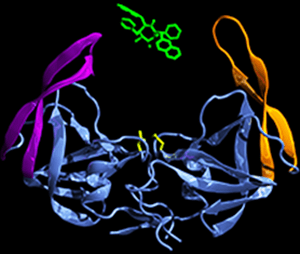
Protein-ligand interaction studies of HIV-1 protease revealed the opening mechanism that allows ligands in to inactivate the enzyme. They give new insight into how multi-drug resistance arises from mutations.
BRIKARD (Coutsias)
Fast searching and sampling of loops & macrocycles
Coutsias’ inverse-kinematics algorithm BRIKARD is 100-fold faster, and gives more accurate ensembles, than other methods that model loops & flexible regions.
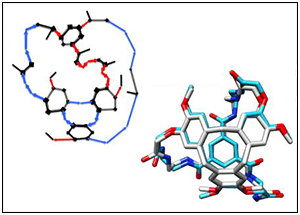
BRIKARD enables exhaustive sampling of the conformations of geometrically complex molecules.
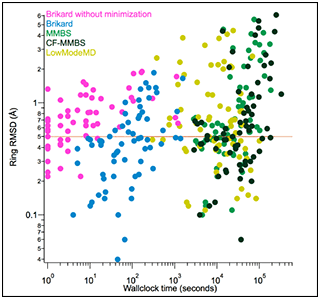
BRIKARD generates low-energy ensembles much faster than other methods.
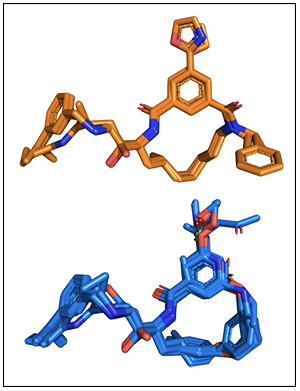
BRIKARD can sample hard-to-access geometries of complex macrocycles.
CLUSPRO (Kozakov)
Computing protein-protein structures
CLUSPRO is among the top protein-protein docking servers, according to CAPRI blind-competition evaluations.
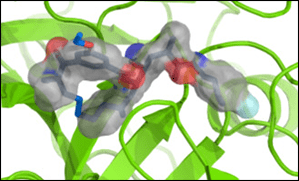
Kozakov’s CLUSPRO – LigTBM builds ligands into protein sites and does virtual screening.
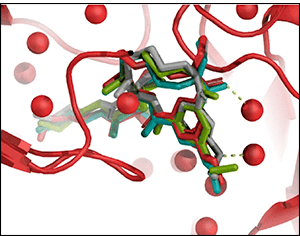
CLUSPRO docks complex ligands with proteins.
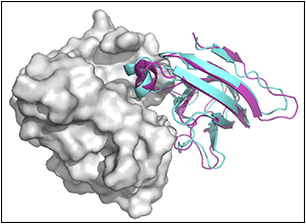
CLUSPRO performs efficient rigid protein-protein docking.
DOCK6 (Rizzo)
Virtual screening and de novo design to identify drug leads
DOCK6 is a powerful tool for small-molecule drug discovery.
The Rizzo lab is a key developer of DOCK, a tool for virtual screening for clinically important targets, leading to compounds with experimentally-verified activity.
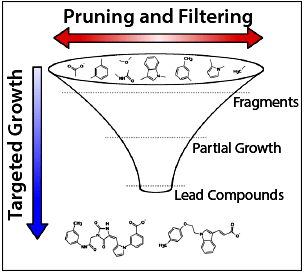
DOCK6 allows tailorable ligand construction, employing fragment libraries derived from millions of drug-like molecules, to design novel tight-binding ligands for protein targets.
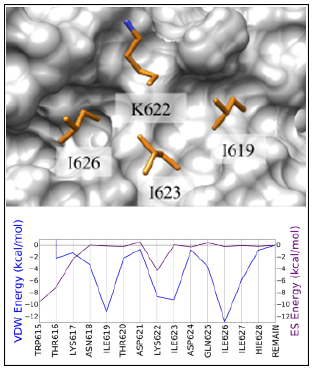
DOCK6 contains powerful similarity-based scoring functions, for finding compounds that mimic energetic and structural signatures in protein sites or interfaces.
MELD (Dill)
An accelerator for Molecular Dynamics (MD) modeling
MELD x MD accelerates MD modeling, often by orders of magnitude. It folds proteins, binds ligands and refines structures and protein-protein dockings.
MELD x MD can fold small proteins from sequence; validated in the CASP competition.
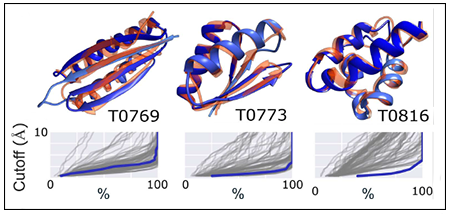
MELD computes affinities of peptides binding to proteins.
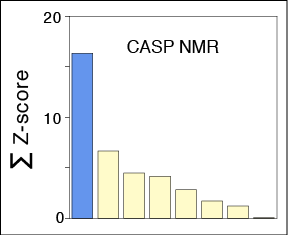
MELD leverages NMR data to determine protein structures; validated in CASP blind tests.
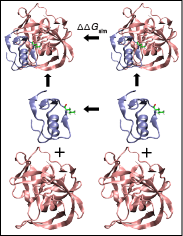
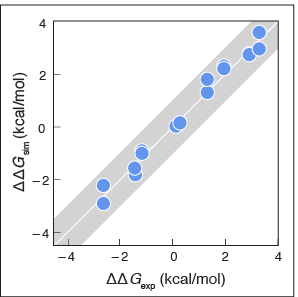
MELD predicts mutational effects on protein-protein binding.
